生物信息分析中的reads是什么
本文最后更新于:2024年6月17日 下午
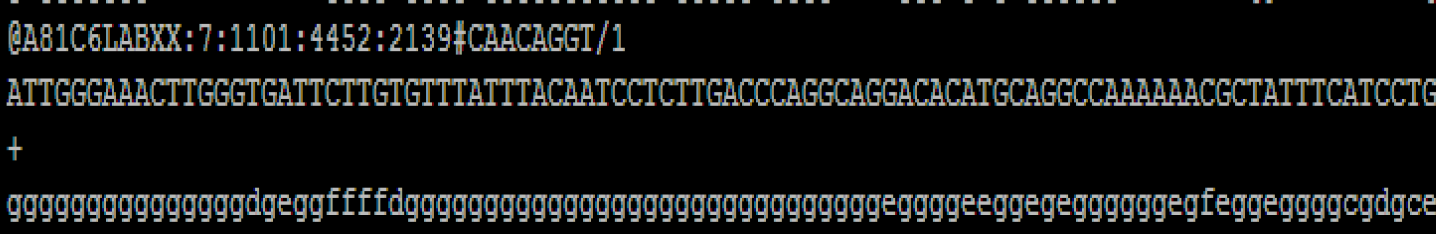 测序得到的原始图像数据经 base calling 转化为序列数据,我们称之为
测序得到的原始图像数据经 base calling 转化为序列数据,我们称之为 raw data 或 raw reads ,结果以 fastq 文件格式存储, fastq 文件为用户得到的最原始文件,里面存储 reads 的序列以及 reads 的测序质量。在 fastq 格式文件中每个 read 由四行描述:
1 | |
- Single-end(SE)测序:1 个 fastq 文件
- Pair-end(PE)测序:2 个 fastq 文件分别存放 read1 和 read2 的数据
每个序列共有 4 行,第 1 行和第 3 行是序列名称(有的 fq 文件为了节省存储空间会省略第三行“+”后面的序列名称);第 2 行是序列;第 4 行是序列的测序质量,每个字符对应第 2 行每个碱基,第 4 行每个字符对应的 ASCII 值减去 64,即为该碱基的测序质量值,比如 h 对应的 ASCII 值为 104,那么其对应的碱基质量值是 40。
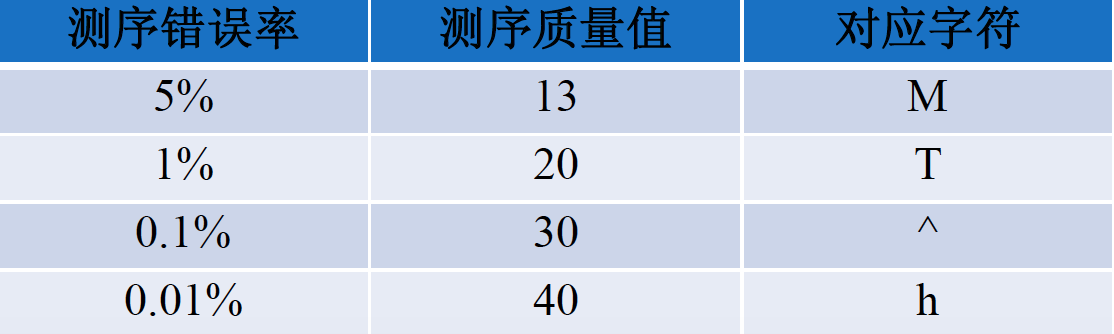
碱基质量值范围为 0 到 40。下表为 Solexa 测序错误率与测序质量值简明对应关系,具体计算公式如下:
1 | |
Solexa 测序错误率与测序质量值简明对应关系:
如果这篇文章对你有帮助,或者想给我微小的工作一点点资瓷,请随意打赏。

微信支付

支付宝
生物信息分析中的reads是什么
https://blog.pangao.vip/生物信息分析中的reads是什么/